3 Doenças tireoidianas no período neonatal
CID-10
E03.0: hipotireoidismo congênito com bócio difuso
E03.1: hipotireoidismo congênito sem bócio
E05.0: tireotoxicose com bócio difuso
E05.5: crise ou “tempestade” tireotóxica
P72.1: hipotireoidismo neonatal transitório
R94.6: resultados anormais de estudos de função tireoidiana
Introdução
Entre as doenças tireodianas no período neonatal, o hipotireoidismo congênito é a doença endócrina congênita mais comum, acometendo 1 em cada 2500 a 3000 recém-nascidos vivos, afeta ambos os sexos na mesma proporção e é a causa evitável mais comum de retardo mental.
Como a maioria dos pacientes é assintomática nas primeiras semanas de vida e, quando presentes, as manifestações clínicas são inespecíficas, os pacientes são identificados pela triagem neonatal biológica (teste do pezinho). O diagnóstico e início do tratamento nas primeiras duas a três semanas de vida garantem o desenvolvimento normal do sistema nervoso central.
Por outro lado, a tireotoxicose congênita é uma situação muito incomum, acomentendo apenas 0,1 a 1% das crianças nascidas de mãe com hipertireoidismo adquirido autoimune e, a despeito de ser uma condição rara na prática do pediatra, as manifestações clínicas podem estar presentes desde o período intrauterino ou ocorrerem somente após o nascimento e , dependendo de sua gravidade, essas crianças podem evoluir com óbito fetal ou nascer prematuramente.
Hipotireoidismo congênito
O hipotireoidismo congênito (HC) é a doença endócrina congênita mais comum, acomete um em cada 2500 a 3000 recém-nascidos vivos, afetando ambos os sexos na mesma proporção e é caracterizado por graus variáveis de deficiência na produção dos hormônios tireoidianos causada por disfunção do eixo tireotrófico.
Etiologias
O HC pode ser classificado didaticamente de acordo com sua duração em permanentes ou transitórios e de acordo a sua etiologia em primários, causado pela incapacidade da tireoide em produzir os hormônios tireoidianos ou centrais, quando há deficiência hipotalâmica ou hipofisária na produção de hormônio tireotrófico (TSH).
Hipotireoidismo congênito primário
Causas permanentes
- Disgenesias tireoidianas:
- Corresponde a aproximadamente 80 a 85% dos casos
- Resultante de alterações no desenvolvimento embriológico da tireoide
- As crianças pode apresentar agenesia tireoidiana completa, hemiagenesia, hipoplasia tireoidiana ou defeitos na migração da tireoide
- Hipotireoidismo isolado ou sindrômico com alterações craniofaciais (atresia de coanas e epiglote bífida); malformações em aparelho genitourinário, cardíacas ou neurológicas.
- São esporádicos na maioria dos casos
- Variantes patogênicas são identificadas em menos de 2% dos casos.
- Disormoniogênese:
- Corresponde aos 10 a 20% restante dos pacientes
- Apresentam defeitos em alguma das etapas da síntese hormonal
- Podem apresentar hipotireoidismo isoladamente com bócio ou não ou hipotireoidismo associado a surdez neurossensorial.
- Outras causas mais incomuns:
- Mutações no receptor de TSH
- Ação de desrreguladores ambientais
Causas transitórias
- Filhos de mãe com hipotireoidismo adquirido autoimume
- Deficiência ou excesso materno de iodo
- Variantes patogênicas em heterozigose dos genes envolvidos no sistema de produção de peróxido de hidrogênio (DUOX2 e DUOXA2)
- Hemangioendotelioma hepático congênito
Hipotireoidismo congênito central
Os hipotireoidismo centrais são bem mais raros que os hipotireoidismos primários, acometendo cerca de um em cada 16000 a 100000 recém-nascidos vivos. A deficiência na secreção de TSH pode ocorrer de forma isolada ou combinada com outras deficiências hipofisárias e estar associada a malformações , principalmente neurológicas (ver Capítulo - Hipopituitarismo)
Apresentação clínica
Cerca de 90% das criancas com hipotireoidismo congênito são assintomáticas no período neonatal. As manifestações clínicas frequentemente ocorrem a partir da segunda ou terceira semana de vida e são inespecíficas (quadro 3.1).
Icterícia neonatal prolongada: 33-59%
Hérnia umbilical: 18-32%
Dificuldades de sucção: 16-35%
Letargia e movimentos lentificados: 14-34%
Hérnia umbilical: 18-32%
Macroglossia: 12-25%
Demora na queda do queda do coto umbilical
Pele fria ou mosqueada: 10-18%
Constipação: 10-18%
Choro rouco: 6-7%
Edema: 3-5%
Hipotermia: 3%
Hipotonia: 3%
Bócio visível: 2-6%
Fonte: adaptado de Grant, DB et al. Arch Dis Child (1992) doi: 10.1136/adc.67.1.87
Diagnóstico
Como a maioria dos pacientes com hipotireoidismo congênito são assintomáticos ou apresentam manifestações clínicas inespecíficas que ocorrem mais tardiamente e o atraso no diagnóstico pode levar a consequências catastróficas e irreversíveis no neurodesenvolvimento, a identificação dos casos suspeitos é feita através da triagem neonatal biológica que deve ser coletada nos primeiros dias de vida.
Os casos identificados como suspeitos devem ser confirmados laboratorialmente, preferencialmente nas primeiras duas semanas de vida.
Triagem neonatal
A coleta da amostra de sangue total em papel-filtro deve ser realizada após 48 horas de vida não ultrapassando o quinto dia de vida.
Os níveis de corte para convocação das crianças ainda não estão definidos, sendo diferentes entre as recomendações do Programa Nacional de Triagem Neonatal (PNTN) do Ministério da Saúde e a Sociedade Brasileira de Pediatria (SBP) (quadro 3.2).
Pelo risco das sequelas no neurodesenvolvimento causadas pelo hipotireoidismo congênito, nós optamos por seguir as recomendações da Sociedade Brasileira de Pediatria em nosso serviço
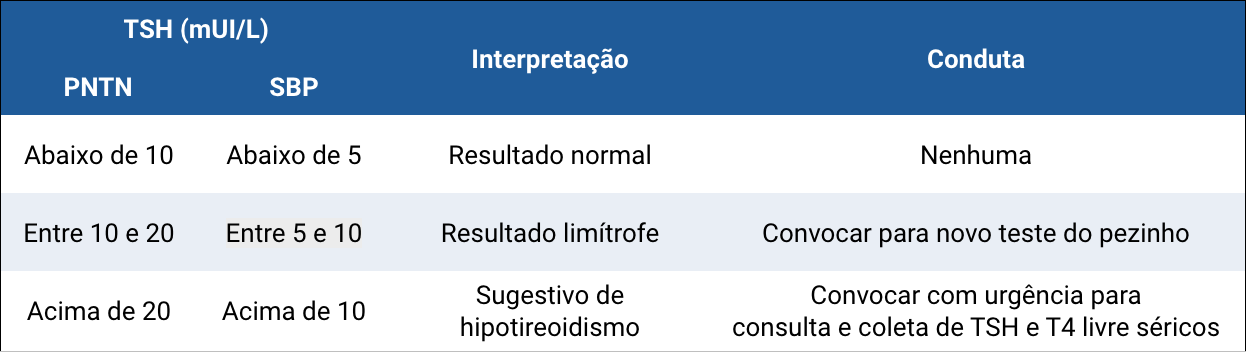
Fonte: adaptado de Guia Prático em Doenças da Tireoide. Sociedade Brasileira de Endocrinologia e Metabologia - isbn: 9786589832041
Como todos os testes de triagem neonatal biológicos, a investigação do hipotireoidismo congênito também está sujeitos tanto a resultados falsamente positivos como falsamente negativos (quadro 3.3).
Resultados falsos-negativos: transfusões de hemoderivados; hipotireoidismo central
Resultados falsos-positivos: prematuridade; baixo peso ao nascer; coleta precoce; medicamentos (dopamina, iodo, glicocorticoides); estresse perinatal (infecção, hipoglicemia); crianças gravemente doentes ou com instabilidade hemodinâmica; filhos de mães tireoidopatas
Confirmação diagnóstica
- Todas as crianças com resultados alterados precisam de confirmação do diagnóstico com avaliação imediata, incluindo história, exame físico e exames laboratoriais (TSH e T4 total ou livre) e que deve ser realizada o mais precoce possível (até a segunda semana de vida).
História clínica
História da moléstia atual: perguntar quanto tempo de vida a criança tinha quando foi realizada a coleta do exame de triagem neonatal e a presença de fatores de risco para resultados falsamente positivos ou negativos (ver Seção 3.3.3.1)
Interrogatório sobre os diversos aparelhos: perguntar ativamente sobre manifestações clínicas que podem estar presentes em crianças com hipotireoidismo (ver Seção 3.3.2) ou aquelas presentes nas formas não isoladas de hipotireoidismo como sopro cardíaco, alterações em exames de imagem cardíacos ou renais identificadas durante internação no berçario, necessidade de ventilação mecânica, manifestações neurológicas como coreoatetose, convulsões, entre outras.
Antecedentes pessoais neonatais: gestação; tipo de parto; idade gestacional; peso, comprimento e perímetro cefálico de nascimento; Apgar; internações e intercorrências no período neonatal.
Antecedentes familiares: consanguinidade; idade e estado de saúde dos pais; estatura dos pais e cálculo da estatura-alvo; informações sobre irmãos; doenças na família, principalmente doenças tireoidianas congênitas ou casos de hipopituitarismo; situação social e financeira
Exame físico
- Identificação de dismorfismos fenotípicos: alterações fenotípicas faciais como cabelos espetados, fenda palatina, epiglote bífida e pseudo-hipertrofia muscular, encontrada em pacientes com hipotireoidismo grave na síndrome de Kocher-Debré-Semelaigne.
A pseudo-hipertrofia afeta os músculos das extremidades, cintura escapular e pélvica, tronco, mãos e pés, sendo mais evidente nos membros, resultando em uma aparência atlética (ver Capítulo - Identificação de dismorfismos fenotípicos)
Antropometria (Seção 2.2.3): peso; comprimento; perímetro cefálico e avaliação dos segmentos somáticos, como a relação entre o segmento superior e inferior.
Baixo ganho ponderoestatural pode ser identificado nos pacientes com diagnóstico tardio.Exame físico geral, segmentar e específico (Seção 2.2.4): manifestações clínicas sugestivas de hipotireoidismo como hipotonia, icterícia, mixedema facial, alargamento de fontanelas, macroglossia e hernia umbilical; avaliação genital e estadiamento puberal
Investigação laboratorial e exames de imagem iniciais
TSH e T4 livre (ou T4 total)
Dosagem de tireoglobulina
Pesquisa de anticorpos tireoidianos (antiperoxidase e antitireoglobulina)
A dosagem de tireoglobulina e a pesquisa de anticorpos tireoidianos podem auxiliar na identificação de crianças com hipotireoidismo permanente por agenesia tireoidiana, que vão apresentar níveis indetectáveis de tireoglobulina ou de casos de hipotireoidismo primário transitório, pela identificação dos anticorpos tranferidos via placentária.
Os valores de TSH para diagnóstico de hipotireodismo congênito primário ainda não estão definidos (quadro abaixo). No nosso serviço, optamos por seguir as recomendações da Sociedade Brasileira de Pediatria.
fazer box no drawio
Além da confirmação laboratorial do hipotireoidismo, recomenda-se a realização de exames de imagem como ultrassonografia e cintilografia de tireoide. Esses exames podem auxiliar na determinação da etiologia e na distinção entre o HC transitório e definitiva.
É importante ressaltar que esses exames não devem retardar o início do tratamento.
Ultrassonografia de tireoide: exame indolor, não invasivo, de baixo custo e sem liberação de radiação ionizante. Pode avaliar a posição, o tamanho e a textura ecogênica da tireoide.
Deve ser realizado, preferencialmente, por um radiologista com experiência em exames da faixa etária pediátrica.Cintilografia tireoidiana: pode ser usada em conjunto com a ultrassonografia para propiciar maior precisão diagnóstica, conseguindo identificar os casos de agenenia, hipoplasia ou ectopia tireodiana de maneira mais confiável e permitir uma melhor avaliação dos casos de hipoplasia tireoidiana.
Crianças com tireoide de localização e tamanho normal para a idade podem ser submetidos a avaliação posterior para descartar HC transitório.
Tratamento e acompanhamento
Tratamento
Tratamento de escolha (hipotireoidismo primário ou central): levotiroxina sódica (LT4)
Iniciar o mais breve possível: idealmente nas primeiras duas semanas de vida ou imediatamente após confirmação através de dosagem sérica.
Dose inicial: depende da gravidade do hipotireoidismo (baseado nos níveis de T4 livre)
Deficiência leve (T4 livre > 0,77 ng/dL): entre 5 e 10 µg/kg/dia.
Deficiência grave (T4 livre < 0,38 ng/dL): entre 10 e 15 µg/kg/dia.
Cuidados no tratamento:
- Administrar a LT4 em jejum (antes da primeira mamada de manhã).
- O comprimido deve ser macerado e diluído em água ou leite materno e administrado com uma colher.
- Não diluir o comprimido na mamadeira.
- Em caso de vômitos até 30 minutos após a administração da LT4: administrar novamente a mesma dose
- Não administrar outros medicamentos (sulfato ferroro, medicamentos anti-epilépticos, carbonato de cálcio ou vitamina D) com a LT4
- Pacientes com deficiência de ACTH devem iniciar o tratamento com LT4 entre três e cinco dias após o início do tratamento com glicocorticoide para minimizar o risco de insuficiência adrenal aguda (Capítulo - Emergências endócrinas).
- Entregar a carta de orientações sobre o tratamento do hipotireoidismo para a família disponível no nosso site.
Acompanhamento ambulatorial
Deve ser feito preferencialmente pela equipe da endocrinologia infantil.
Na impossibilidade de atendimento breve, o pediatra deve fazer este acompanhamento até que o paciente inicie seu acompanhamento com nossa equipe.
Primeira consulta ambulatorial:
\scriptsize{\bullet} Quando: três a quatro semanas após introdução do tratamento
\scriptsize{\bullet} Avaliação clínica: presença de manifestações clínicas sugestivas de hipotireodismo, velocidade de crescimento e ganho ponderal e desenvolvimento neuropsicomotor
\scriptsize{\bullet} Avaliação laboratorial: TSH e T4 livre
Devem estar dentro do intervalo de normalidade para a idade.
Pacientes com hipotireoidismo central: o controle é feito somente com o T4 livre.
Ajustes no tratamento:
\scriptsize{\bullet} No nosso serviço, a dose é escalonada em 12,5 µg por vez.
\scriptsize{\bullet} Níveis muito diminuídos ou suprimidos de TSH ou sinais de tireotoxicose: reduzir a dose em 12,5 µg
\scriptsize{\bullet} Níveis de TSH ainda estiverem elevados: aumentar a dose em 12,5 µg, mesmo que os níveis de T4 livre estejam normais, o paciente esteja assintomático e apresente ganho ponderal e/ou crescimento adequados
\scriptsize{\bullet} Após ajuste da dose, o paciente deve ser reavaliado novamente em três a quatro semanas
Seguimento ambulatorial:
\scriptsize{\bullet} O intervalo entre as consultas varia conforme a idade do paciente
\scriptsize{\circ} Até 6 meses: mensal
\scriptsize{\circ} Entre 6 meses e 3 anos: entre dois e três meses
\scriptsize{\circ} Após os 3 anos de idade (até o término do crescimento): entre 3-6 meses
Investigação de outras malformações:
\scriptsize{\bullet} Teste de triagem auditiva: todas as crianças
\scriptsize{\bullet} Eletrocardiografia e ecocardiografia: crianças com sopro cardíaco ou outras manifestações de cardiopatias congênitas
Encaminhar para avaliação com cardiologista se for necessário.
\scriptsize{\bullet} Acompanhamento multidisciplinar e multiprofissional especializado: crianças com alterações neurocognitivas devem ser encaminhadas para estimulação do desenvolvimento motor e até mesmo um plano educacional personalizado.
Tireotoxicose congênita
Os hipertireodismos congênitos são situações muito incomuns na vida do pediatria e do neonatologista e podem acomenter entre 0,1 a 1% das crianças filhas de mãe com hipertireoidismo adquirido autoimune, o que representa cerca de 1 em cada 25000 a 50000 nascidos vivos.
Muito mais raro ainda é o hipertireodismo causado por mutação ativadora do receptor de TSH, que ocorre na síndrome de McCune-Albright.
Apresentação clínica
As manifestações clínicas podem estar presentes desde o período intrauterino, com tireotoxicose fetal ou podem ocorrer somente após o nascimento, no período neonatal.
As manifestações clínicas podem estar presentes desde o período intrauterino, com tireotoxicose fetal ou podem ocorrer somente após o nascimento, no período neonatal
Tireotoxicose fetal
Retardo de crescimento intrauterino
Oligodrâmnio
Taquicardia
Bócio
Prematuridade
Tireotoxicose neonatal
- Manifestações metabólicas: aumento no apetite, baixo ganho ponderal, hipertermia e sudorese excessiva
Manifestações cardiovasculares: taquicardia, hipertensão arterial, instabilidade hemodinâmica e hipertensão pulmonar
Manifestações neurológicas: irritabilidade, sono agitado, fechamento prematuro das fontanelas, microcefalia
Manifestações respiratórias: taquipneia e desconforto respiratório, necessitando de suporte ventilatório
Outras manifestações: diarreia, hepatomegalia, trombocitopenia e icterícia
Diagnóstico
Todo paciente com risco para doença de Graves neonatal (quadro 3.7) ou evidências de tireotoxicose fetal exigem avaliação imediata, incluindo história, exame físico e exames laboratoriais.
Níveis de TRAb elevados durante gravidez
Doença de Graves materna ativa
Níveis de TRAb não avaliados durante gravidez
Uso de drogas antitireoidianas durante 2º ou 3º trimestre
Tireotoxicose durante 2º ou 3º trimestre de gestação
Gestação anterior com recém-nascido apresentando doença de Graves
História familiar de mutação no gene do receptor de TSH
Fonte: elaborado pelo autor
História clínica
Interrogatório sobre os diversos aparelhos: se a família estiver presente na unidade neonatal, perguntar sobre manifestações clínicas tireotoxicose
Antecedentes pessoais neonatais: gestação; intercorrências durante a gestação como oligoâmnio, RCIU, achado ultrassonográfico de taquicardia e/ou bócio fetal; tipo de parto; idade gestacional; peso, comprimento e perímetro cefálico de nascimento; Apgar e intercorrências no período neonatal
Antecedentes familiares: consanguinidade; idade e estado de saúde dos pais; informações sobre irmãos; doenças ou mortes na família; informações sobre o diagnóstico e tratamento do hipertireoidismo materno (etiologia do quadro, medicamentos utilizados e dose atual, adesão ao tratamento e níveis mais recentes de anticorpo antirreceptor de TSH (TRAb) nos casos de doença de Graves.
Exame físico
Antropometria (Seção 2.2.3): peso; comprimento; perímetro cefálico e avaliação dos segmentos somáticos (relação entre o segmento superior e segmento inferior)
As crianças com tireotoxicose fetal e neonatal podem apresentar baixo ganho ponderal ou até perda ponderalExame físico geral, segmentar e específico (Seção 2.2.4): manifestações clínicas sistêmicas sugestivas de hipertireoidismo e exame da tireode.
Investigação laboratorial inicial
Todas as crianças com evidências de tireotoxicose ou fatores de risco para doença de Graves neonatal (quadro 3.7) devem realizar investigação laboratorial (quadro 3.8).
Crianças assintomáticas
Sangue do cordão umbilical: TSH e T4 livre; TRAb, TPO-Ab e TG-Ab ou
Sangue periférico do recém-nascido: pesquisa do TRAb
Crianças sintomáticas
Internar em unidade de terapia intensiva neonatal
Coletar exames: TSH e T4 livre; TRAb, TPO-Ab e TG-Ab (sangue periférico)
Iniciar o tratamento para o mais precoce possível
Tratamento e acompanhamento ambulatorial
Crianças assintomáticas
Local de observação: alojamento conjunto ou unidade de cuidados intermediários
Entre o 1º e 3º dia de vida: avaliação clínica com atenção à presença de sinais de tireotoxicose
Entre o 3º e 5º dia de vida:
\scriptsize{\bullet} Assintomáticas e TRAb negativo: alta hospitalar, sem necessidade de novas coletas de exame e com seguimento ambulatorial de puericultura normal
\scriptsize{\bullet} TRAb positivo ou sem resultado: manter avaliação clínica e coletar TSH e T4 livre em sangue periférico.
Crianças sintomáticas
O tratamento de primeira linha inclui o uso de tionaminas associada ou não aos bloqueadores de receptores beta-adrenérgicos
Normalmente, os medicamentos de primeira linha conseguem controlar as manifestações de tireotoxicose
Nos casos de tempestade tireoidiana ou de difícil controle metabólico, podem ser utilizados medicamentos de segunda linha como o iodeto de potássio e glicocorticoides.
Local de observação: unidade de terapia intensiva neonatal
A. Drogas antitireoidianas
Diminuem a produção dos hormônios tireoidianos e inibem a proliferação folicular.
\scriptsize{\bullet} Droga de escolha: metimazol (MMI)
\scriptsize{\bullet} Dose inicial: 0,2 a 0,5 mg/kg/dia, via oral ou sonda gástrica
A dose diária deve ser dividida em duas doses (a cada 12 horas)
\scriptsize{\bullet} Efeitos adversos: rash cutâneo; urticária; dor abdominal; diarreia; náuseas ou vômitos; agranulocitose e/ou leucopenia e hepatite medicamentosa
B. Bloqueadores de receptores beta-adrenérgicos
Diminuem os efeitos adrenérgicos dos hormônios tireoidianos
\scriptsize{\bullet} Propranolol
\scriptsize{\circ} Dose inicial: 1 a 2 mg/kg/dia, via oral ou sonda gástrica
A dose diária deve ser dividida em 2 a 3 doses (a cada 8 ou 12 horas)
\scriptsize{\circ} Efeitos adversos: hipoglicemia grave, bradicardia e hipotensão arterial.
Em alguns pacientes, pode ser necessário tratamento específico para insuficiência cardíaca.
\scriptsize{\bullet} Atenolol
\scriptsize{\circ} Dose inicial: 0,5 mg/kg/dia, via oral ou sonda gástrica, uma vez ao dia
Se necessário, a dose pode ser aumentada e administrada a cada 12 horas.
Tem uma ação mais cardiosseletiva que o propranolol.
\scriptsize{\circ} Efeitos adversos: os mesmos do propranolol
C. Iodeto de potássio
Inibe a secreção dos hormônios tireoidianos e inibem a conversão extratireoidiana de T4 em T3.
\scriptsize{\bullet} Dose inicial: 50 mg (1 gota), via oral ou por sonda gástrica, uma vez ao dia
Pode ser administrada até duas vezes ao dia.
D. Glicocorticoides
Inibem a conversão extratireoidiana de T4 em T3.
\scriptsize{\bullet} Prednisolona
\scriptsize{\circ} Dose inicial: 1 a 2 mg/kg/dia, administrada por via oral, uma vez ao dia.
Alta hospitalar
Os pacientes só devem receber alta hospitalar após melhora do quadro clínico e estabilização metabólica.
Antes da alta, a família deve ser orientada a procurar atendimento médico imediatamente no caso da criança apresentar efeitos adversos graves das tionaminas como colúria, acolia fecal, icterícia, dores abdominais, artralgias ou febre
Acompanhamento ambulatorial
Primeira consulta ambulatorial
\scriptsize{\bullet} Quando: 7 e 14 dias após a alta hospitalar
\scriptsize{\bullet} Avaliação clínica: presença de manifestações clínicas sugestivas de hipertireodismo, velocidade de crescimento e ganho ponderal e desenvolvimento neuropsicomotor neuropsicomotor
Segunda consulta
\scriptsize{\bullet} Quando: 30 dias após a alta com avaliação clínica e laboratorial
\scriptsize{\bullet} Avaliação clínica
\scriptsize{\bullet} Exames laboratoriais: TSH, T3 total e T4 livre; TGO, TGP e gama-GT e hemograma
\scriptsize{\circ} TSH: próximo do limite inferior da normalidade
\scriptsize{\circ} T3 total e T4 livre: dentro do intervalo de normalidade para a idade.
\scriptsize{\circ} TGO e TGP: até 3 vezes o limite superior de normalidade
\scriptsize{\circ} Hemograma completo: contagem de leucócitos e neutrófilos normal
Ajustes no tratamento
\scriptsize{\bullet} No nosso serviço, a dose é escalonada em 10 a 20% por vez.
\scriptsize{\bullet} Níveis suprimidos de TSH ou sinais de tireotoxicose: aumentar a dose em 10 a 20%, mesmo que o paciente esteja assintomático e ganho ponderal e/ou crescimento adequados e níveis de T3 e T4 livre estejam normais
\scriptsize{\bullet} Níveis de TSH acima do limite inferior ou elevados: reduzir a dose 10 a 20%, mesmo que níveis de T3 e T4 livre estejam normais
Seguimento ambulatorial
\scriptsize{\bullet} Intervalo entre as consultas: entre três e seis semanas e inclui tanto avaliação clínica como laboratorial.
\scriptsize{\bullet} Hormônios tireoidianos normalizam ao redor de 4 a 8 semanas
\scriptsize{\bullet} TSH normaliza ao redor de 2 a 3 meses
Para mais detalhes sobre o tratamento e acompanhamento, ler o Capítulo - Hipertireodismos adquiridos.
Bibliografia
van Trotsenburg P et al. Congenital Hypothyroidism: A 2020–2021 Consensus Guidelines Update—An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrino- logy and the European Society for Endocrinology. Thyroid. 2021; 31(3): 387-419.2. doi: 10.1089/thy.2020.0333
Cherella CE, Wassner AJ. Update on congenital hypothyroidism: Current Opinion in Endocrinology & Diabetes and Obesity. 2020; 27(1): 63-9.3.
Peters C, van Trotsenburg ASP, Schoenmakers N. Congenital hypothyroidism: update and perspectives. European Journal of Endocrinology. 2018; 179(6):R297–317.
Wassner AJ. Congenital Hypothyroidism. Clin Perinatol. 2018; 45(1): 1–18.5. doi: 10.1016/j.clp.2017.10.004
Conitec. Protocolo Clínico e Diretrizes Terapêuticas do Hipotireoidismo Congênito. Brasília/DF: Ministério da Saúde; 2021. (Protocolo Clínico e Diretrizes Terapêuticas)
Sociedade Brasileira de Pediatria. Hipotireoidismo Congênito: Triagem Neonatal. Sociedade Brasileira de Pediatria; 2018 p. 12. (Documento Científico de Endocrinologia).
Léger J, Carel JC. Diagnosis and management of hyperthyroidism from prenatal life to adolescence. Best Pract Res Clin Endocrinol Metab. 2018;32(4):373–86. doi: 10.1016/j.beem.2018.03.014
Ogilvy-Stuart AL. Neonatal thyroid disorders. Arch Dis Child. 2002;87(3):165F-171. doi: 10.1136/fn.87.3.F165
Samuels SL, Namoc SM, Bauer AJ. Neonatal Thyrotoxicosis. Clin Perinatol. 2018;45(1):31–40. doi: 10.1016/j.clp.2017.10.001